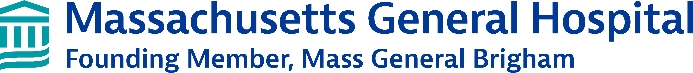

Rapid Translation of Human Neuron Screen Hit from the Lab to Clinical Trial A proposal from the Wainger Lab, Brian Wainger, MD, PhD
Wainger Therapeutic proposal
ALS and Frontotemporal Disorder are likely caused by multiple pathological processes occurring at different locations on motor neurons. In particular, evidence suggests that distinct mechanisms underly neuronal toxicity in the cell body, the control center of the neuron and the axons that project outwards from the cell body. To address these separate toxicities and leverage our improved tools and scale, we used human stem cell-derived neurons and generated over
400 million neurons expressing a nuclear blue fluorescent protein signal and a membrane-bound red fluorescent signal from within the cell. These fluorescence markers allow us to more precisely monitor cell body and neurite degeneration. We then use these calculations in screens to identify new potential treatments.
The group has developed screens using these human neurons to identify drugs that target multiple molecular mechanisms of ALS at the same time. Two independent screens identified a family of compounds that includes an FDA-approved drug.
We are proposing a four-pronged approach to move as quickly as possible to advance the potential of these results.
1) We will perform a six-month, open label clinical trial of the drug
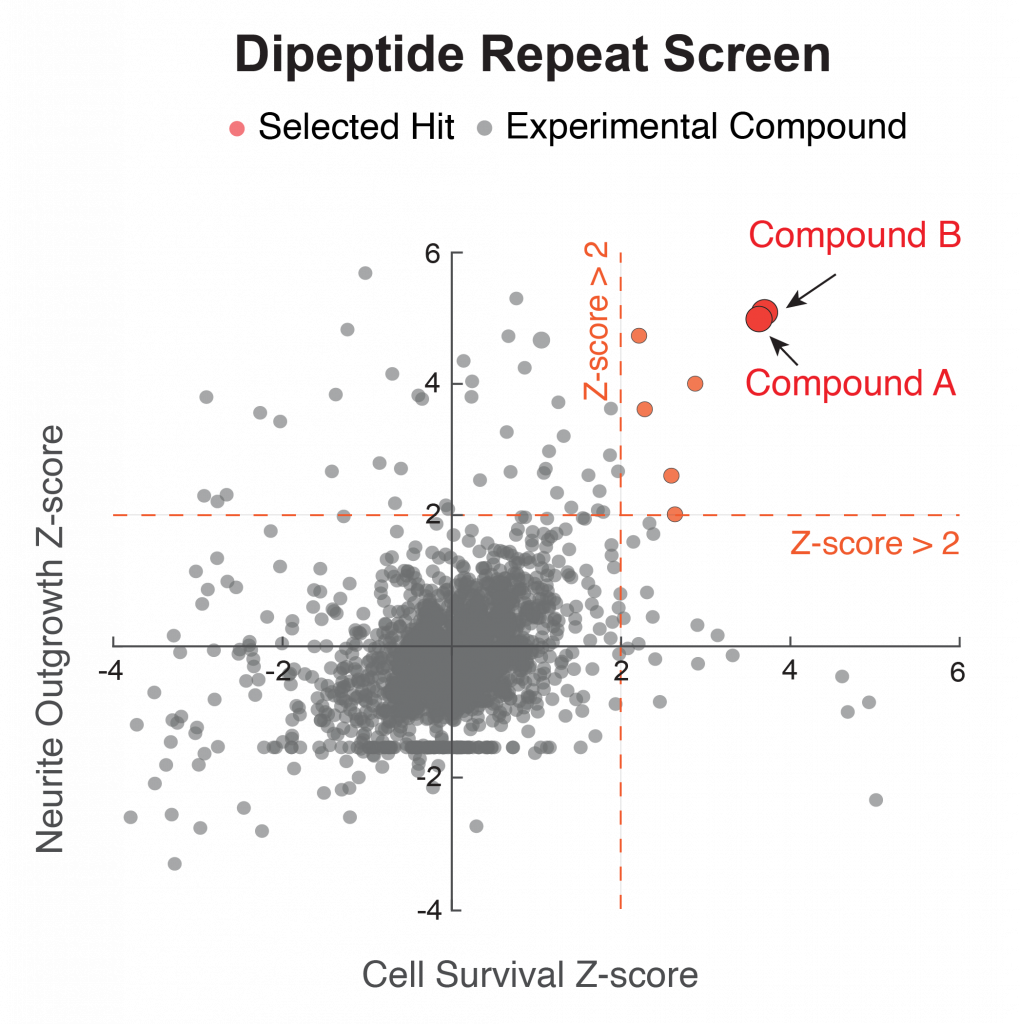
Figure 1. Screen using dual readouts of cell survival and neurite outgrowth following toxic C9orf72 dipeptide repeats (DPR) reveals two hits within the same pharmacological family (red), one of which is an FDA-approved drug. Neurons labeled with nuclear blue fluorescent and membrane-bound red fluorescent markers can be used to track neurite retraction and survival. A 2,000 compound screen identifies hit compounds that improve both neuronal survival and neurite length after DPR treatment.
(compound A) in approximately 15 people with C9orf72ALS , including determining CSF levels, DPR levels and neurofilament levels (a biomarker for ALS).
2) We will perform validation in a C9orf72 mouse model. Despite limitations of mouse models for ALS, we believe that reduction of DPR toxicity in an animal model will help build critical, long-term support for the program.
3) We will perform experiments in our lab to better understand the mechanistic target of the drug’s benefit in ALS/FTD. We will use genetic and pharmacological approaches, and this work will also open the door to antisense oligonucleotide and other genetic-based treatment modalities.
4) One limitation is that the current drug (Compound A) has limited entry to the central nervous system (CNS), although separate evidence suggests that the neuroinflammation present in ALS/FTD may enhance its CNS entry. We have begun work with a medicinal chemistry team to optimize the CNS entry of the compound. We already have identified core structural components of the molecule necessary for its activity, including one compound with efficacy at only 1 nM and improved CNS access. The goal will be to develop an improved molecule with greater CNS entry and stronger potency.
We performed an initial screen using a toxic dipeptide repeat (DPR), which is a primary mechanism of toxicity in C9orf72 ALS/FTD. While genetic screens have provided insights into the mechanisms of DPR toxicity by identifying genetic modifiers, no clear therapeutic candidates have emerged. To identify compounds with potential therapeutic benefit, we performed a high-throughput phenotypic screen of a chemical library of about 2,000 compounds against poly(GR) toxicity in human stem cell-derived neurons. We identified hits that were protective for reducing both cell body and axonal toxicity. The top hits included two compounds within the same chemical
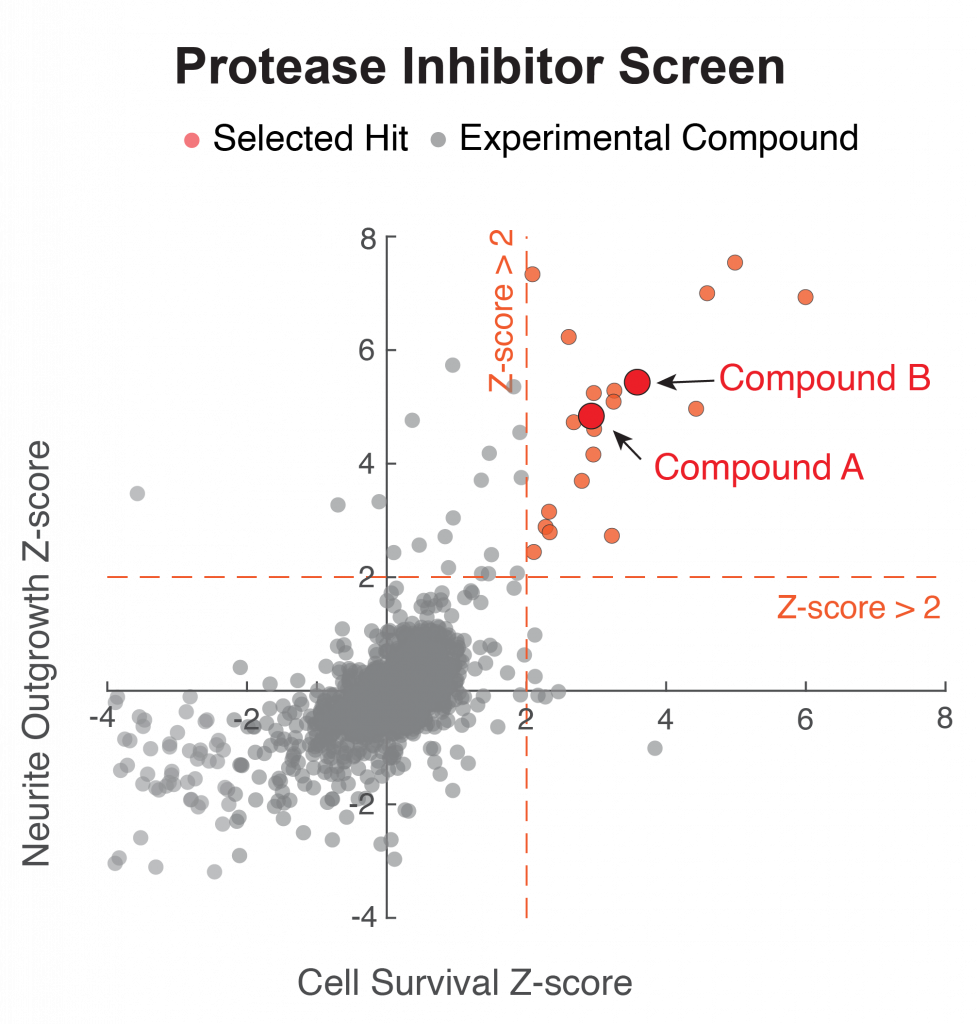
Figure 3. An independent screen based on toxicity of the protease inhibitor bortezomib identifies the same two hits (red).
family, including one that is FDA-approved (Figure 1). We validated the hits and in dose response curves found that concentrations as low as 10 nM led to substantial reduction in poly(GR) toxicity (Figure 2). Moreover, the compound is protective against both poly(GR) and poly(PR), the sense and antisense DPRs thought to be most toxic in C9orf72 ALS/FTD.
We next performed a second screen using the protease inhibitor bortezomib, which is used to model TDP-43 mislocalization, with the same setup. In this completely independent screen, we also identified the same two compounds as hits (Figure 3). Notably, treatment reversed
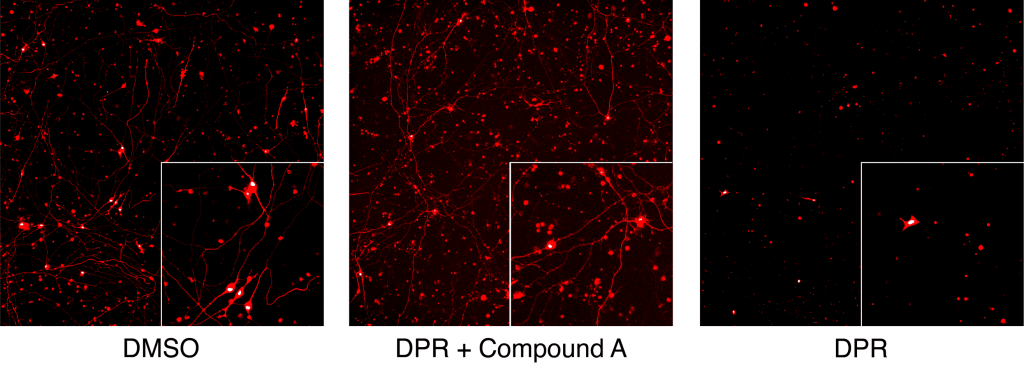
Figure 2. Demonstration of the benefit in reducing toxicity of C9orf72 dipeptide repeat (DPR) of the FDA approved drug on neuronal cell bodies and neurites. Compound A is the FDA approved drug, applied at nanomolar concentration.
pathological TDP-43 mislocalization at sub-micromolar concentrations (Figure 4). This is particularly encouraging, because the cytoplasmic mislocalization and aggregation of TDP-43 is the main neuropathological feature present in 98% of ALS and half of FTD.
Thus, we propose to accelerate the translational development of hits from this screen, including in a clinical trial of the FDA approved drug, mouse studies of C9orf72 ALS, identification of genetic mechanistic targets, and development of improved compounds based on the hits.